Vasculitis: Understanding Autoimmune Inflammation of Blood Vessels
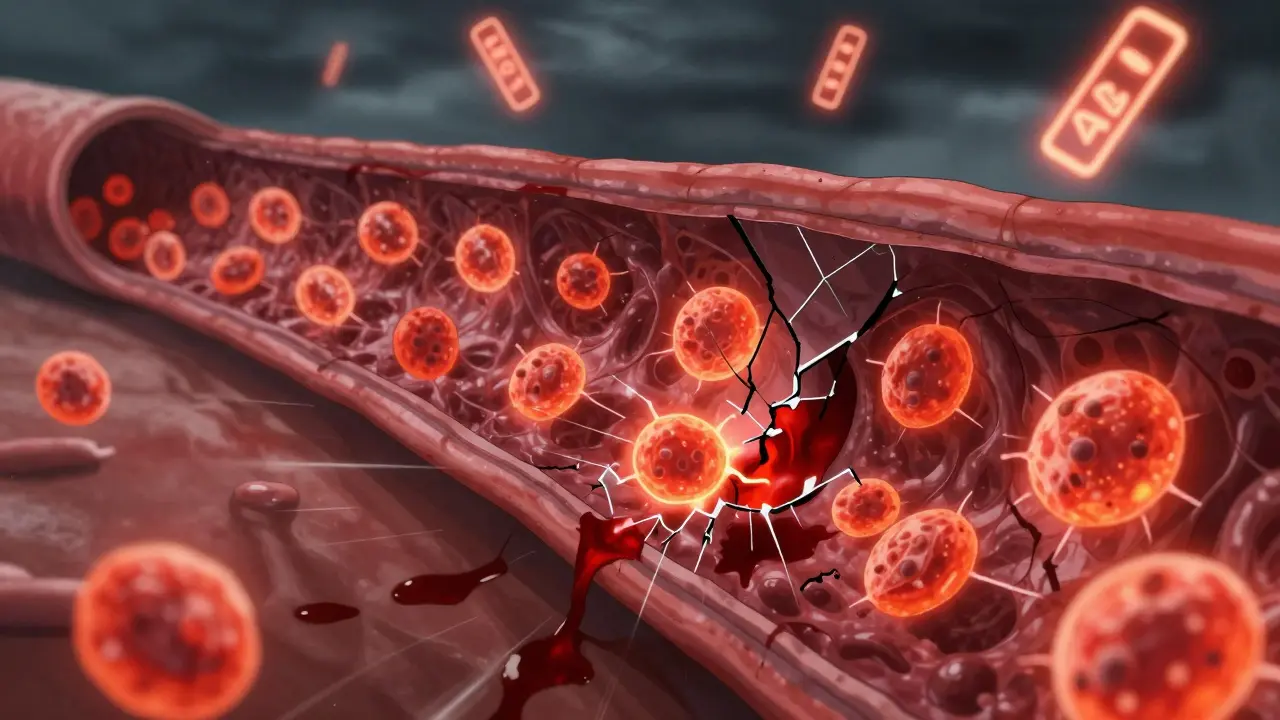
Vasculitis is not one disease-it’s a group of rare autoimmune disorders where the immune system turns against the body’s own blood vessels. Instead of protecting you, it attacks the walls of arteries, veins, and capillaries, causing inflammation, narrowing, blockages, or even ruptures. This can starve organs of oxygen and lead to serious damage-or death-if left untreated. What makes vasculitis tricky is that its symptoms mimic common illnesses: fatigue, joint pain, fevers, rashes, or numbness in fingers and toes. Many people wait months before getting the right diagnosis.
How Vasculitis Attacks Your Blood Vessels
Think of your blood vessels as highways for oxygen and nutrients. In vasculitis, the immune system sends inflammatory cells-like neutrophils and lymphocytes-straight into the vessel walls. These cells tear through the layers: the inner lining (intima), the middle muscle layer (media), and the outer connective tissue (adventitia). As they do, the vessel swells, weakens, or scars shut. In some cases, it bulges into an aneurysm. In others, it closes completely, cutting off blood flow to the kidneys, lungs, skin, or nerves.
The damage depends on which vessels are targeted. Large vessels like the aorta or temporal arteries can lead to strokes or vision loss. Medium vessels feeding the heart or intestines can cause heart attacks or bowel perforation. Small vessels in the kidneys or lungs may show no symptoms until kidney failure or coughing up blood occurs. That’s why vasculitis is often called a silent killer-it can be advancing without obvious signs.
Types of Vasculitis, Classified by Vessel Size
Doctors group vasculitis by the size of the blood vessels most affected. This helps predict symptoms, guide testing, and choose treatment.
- Large-vessel vasculitis: Affects the aorta and its major branches. Giant cell arteritis (GCA) hits people over 50, often causing severe headaches, jaw pain when chewing, and sudden vision loss. Takayasu arteritis mostly affects young women and can cause weak pulses in the arms or high blood pressure from narrowed arteries.
- Medium-vessel vasculitis: Targets arteries like those in the kidneys, liver, or intestines. Polyarteritis nodosa (PAN) causes abdominal pain, nerve damage, and skin ulcers. Kawasaki disease, seen mostly in children under 5, inflames coronary arteries and can lead to heart aneurysms if not treated quickly with IVIG and aspirin.
- Small-vessel vasculitis: Most common and often linked to autoantibodies called ANCA. Granulomatosis with polyangiitis (GPA) affects sinuses, lungs, and kidneys, sometimes causing bloody nasal discharge or coughing blood. Microscopic polyangiitis (MPA) hits kidneys and lungs hard, often without visible skin signs. Eosinophilic granulomatosis with polyangiitis (EGPA), formerly Churg-Strauss, starts with asthma and allergies, then attacks nerves and organs.
There are others too-like Behçet’s disease, which causes mouth sores, eye inflammation, and genital ulcers, or Buerger’s disease, which is tied directly to smoking and destroys small arteries in hands and feet.
Diagnosis: Why It Takes So Long
Most people see four or five doctors before getting diagnosed. Why? Because symptoms overlap with infections, arthritis, flu, or even stress. A person with fatigue, weight loss, and numb hands might be told they have fibromyalgia-or just need more sleep.
But there are clues. Blood tests often show high ESR (over 50 mm/hr) or CRP (over 5 mg/dL), both markers of inflammation. The big red flag is ANCA antibodies. In GPA, c-ANCA targeting proteinase-3 shows up in 80-90% of cases. In MPA, p-ANCA targeting myeloperoxidase is common.
Still, the gold standard is a tissue biopsy. If you have a skin rash, a small sample can show leukocytoclastic vasculitis-white blood cell debris around vessels. If your kidneys are involved, a kidney biopsy reveals crescent-shaped damage in the glomeruli. Imaging like CT or PET scans can spot inflamed aorta or other large vessels.
The American College of Rheumatology warns: "Vasculitis may seriously affect kidneys without many symptoms initially." That’s why even if you feel okay, doctors check urine for blood or protein and test kidney function. Missing this can mean irreversible damage.
Treatment: From Steroids to Targeted Drugs
Treatment depends on severity and which organs are involved. For mild cases-like small skin rashes-low-dose steroids may be enough. But for life-threatening cases, you need aggressive therapy.
First-line treatment for severe vasculitis usually combines high-dose prednisone (0.5-1 mg/kg daily) with either cyclophosphamide or rituximab. These drugs shut down the overactive immune response. Once remission is reached, maintenance therapy kicks in with methotrexate, azathioprine, or continued rituximab every 6 months for 18-24 months.
For giant cell arteritis, steroids are still the mainstay-but now tocilizumab, an IL-6 blocker, is approved as an add-on. It cuts steroid use by half, reducing side effects like bone loss and diabetes.
One of the biggest advances in recent years is avacopan, approved by the FDA in 2021. It blocks a protein called C5a that drives inflammation. In the ADVOCATE trial, patients on avacopan plus low-dose steroids had the same remission rates as those on high-dose steroids-but they took 2,000 mg less of prednisone over a year. That’s huge for avoiding weight gain, mood swings, and fractures.
For EGPA, mepolizumab (an anti-IL-5 drug) has shown a 50% drop in relapses. And for Buerger’s disease, the only treatment that works? Quitting tobacco. No drug, no surgery-just stop smoking, or the disease will keep coming back.
Prognosis: Can You Live a Normal Life?
With early treatment, 80-90% of people with ANCA-associated vasculitis go into remission. But here’s the catch: about half will relapse within five years. That’s why lifelong monitoring is critical. Blood tests, urine checks, and occasional imaging are part of the routine.
The Five Factor Score helps predict survival in PAN. If you have no major organ damage, your 5-year survival is 95%. But if you have kidney failure, heart issues, or gut damage, it drops to 50%. That’s why catching it early isn’t just helpful-it’s life-saving.
Children with Kawasaki disease have a good outlook if treated within 10 days. But if coronary arteries swell, they need lifelong cardiology follow-up. Some adults with GCA may need steroids for years, but many can taper off safely with newer drugs like tocilizumab.
Still, vasculitis can be fatal. If it blocks blood flow to the brain, heart, or kidneys, death can happen fast. That’s why patients are told: "Don’t ignore new numbness, chest pain, or blood in your urine."

What’s Next? Research and Hope
Scientists are now looking for better ways to predict flares. Blood tests for BAFF (B-cell activating factor) and urinary MCP-1 might one day tell doctors when a relapse is coming-before symptoms start.
Clinical trials are testing abatacept for giant cell arteritis and new biologics for rare forms. The goal? Move away from broad immunosuppression and toward precision medicine-targeting only the broken parts of the immune system.
Patients are also forming support networks. Online groups share tips on managing steroid side effects, finding rheumatologists who specialize in vasculitis, and navigating insurance for expensive biologics.
Common Symptoms to Watch For
Don’t dismiss these-especially if they come together:
- Purple or red spots, bumps, or bruises on legs or feet
- Joint pain or muscle aches without injury
- Shortness of breath or coughing up blood
- Stomach pain, nausea, or bloody stools
- Numbness, tingling, or weakness in hands or feet
- Headaches, jaw pain, or sudden vision changes (especially over 50)
- Fever, night sweats, or unexplained weight loss
If you have two or more of these and they last more than a few weeks, see a rheumatologist. Don’t wait for your primary care doctor to say "it’s just aging" or "stress."
Is vasculitis contagious?
No, vasculitis is not contagious. It’s an autoimmune disorder, meaning your own immune system attacks your blood vessels. You can’t catch it from someone else, and you can’t give it to others. It’s not caused by infection, though infections can sometimes trigger it in people who are genetically predisposed.
Can vasculitis be cured?
There’s no permanent cure yet, but most people can achieve long-term remission. With the right treatment, symptoms disappear and organ damage stops progressing. Many patients live normal, active lives for decades. The challenge is relapse-about half of those with ANCA-associated vasculitis will have another flare. That’s why ongoing monitoring and adherence to maintenance therapy are essential.
What triggers vasculitis?
The exact trigger isn’t known, but it’s thought to be a mix of genetics and environment. Some people have genes that make their immune system more likely to go haywire. Infections, certain medications, or exposure to toxins might act as a spark. Smoking is a major trigger for Buerger’s disease. In some cases, vasculitis develops after another autoimmune condition like lupus or rheumatoid arthritis.
Do I need to take steroids forever?
Not necessarily. Steroids like prednisone are used to quickly control inflammation, but they’re not meant for lifelong use because of side effects-weight gain, diabetes, bone loss, cataracts. Most people taper off steroids within months, especially with newer drugs like rituximab, avacopan, or tocilizumab. These allow you to reduce or even eliminate steroids over time while keeping the disease in check.
Can children get vasculitis?
Yes. Kawasaki disease is the most common form in children under 5. It causes fever, rash, swollen hands and feet, red eyes, and cracked lips. If untreated, it can lead to coronary artery aneurysms in 20-25% of cases. But with early IVIG and aspirin, most children recover fully. Other forms like microscopic polyangiitis are rare in kids but can happen.
How often should I see a rheumatologist?
When first diagnosed and during active treatment, you’ll likely see your rheumatologist every 2-4 weeks. Once in remission, visits may drop to every 3-6 months. Blood tests (ESR, CRP, ANCA, kidney function) and urine checks are done at each visit. If you’re on a biologic like rituximab, you’ll need regular infusions every 6 months. Always report new symptoms-even small ones-right away.
Next Steps: What to Do If You Suspect Vasculitis
If you’re experiencing unexplained symptoms that match those listed above, start by asking your doctor for basic blood work: ESR, CRP, kidney function, and ANCA testing. If results are abnormal or symptoms persist, ask for a referral to a rheumatologist. Don’t wait. Early treatment can prevent permanent damage.
Keep a symptom journal: note when pain started, what triggers it, and how it changes. Bring it to your appointment. Bring a list of all medications, including supplements. And don’t be afraid to get a second opinion-if you feel dismissed, find a specialist who listens.
Vasculitis is rare, but it’s real. And with modern treatments, it’s manageable. You don’t have to live in fear. You just need the right diagnosis-and the right care team.



chandra tan
January 10, 2026 AT 02:33Bradford Beardall
January 11, 2026 AT 06:19lisa Bajram
January 12, 2026 AT 07:26Paul Bear
January 13, 2026 AT 14:46McCarthy Halverson
January 15, 2026 AT 05:29Aurora Memo
January 16, 2026 AT 17:12neeraj maor
January 17, 2026 AT 12:28Michael Marchio
January 18, 2026 AT 03:23Ashlee Montgomery
January 19, 2026 AT 08:07Ritwik Bose
January 20, 2026 AT 20:47Dwayne Dickson
January 20, 2026 AT 22:31Jake Kelly
January 21, 2026 AT 08:40